Defekty w kryształach wpływają na strukturę całego materiału. Fizycy na przykładzie kryształu węglika krzemu pokazali, że nawet tak wymagające obliczeniowo defekty można z powodzeniem badać z dokładnością atomową za pomocą umiejętnie skonstruowanego modelu.
Defekty w kryształach, zwłaszcza dyslokacje krawędziowe o charakterze długich uskoków, wpływają na strukturę całego materiału i modyfikują jego podstawowe właściwości, redukując możliwości zastosowań. Teoretycy zajmujący się modelowaniem kryształów od dawna próbowali więc przewidywać wpływ takich niedoskonałości na właściwości fizyczne materiałów.
Modele, bazujące na wynikach różnych eksperymentów, opisywały zmiany podstawowych własności materiału bez wyjaśniania rzeczywistych przyczyn i skutków zaistniałych zjawisk. Dopiero nowy model węglika krzemu (SiC), zbudowany przez fizyków z Instytutu Fizyki Jądrowej Polskiej Akademii Nauk (IFJ PAN) w Krakowie, pozwolił zademonstrować, że już dziś można modelować kryształy nawet z tak złożonymi defektami jak dyslokacje krawędziowe i wyjaśniać ich cechy procesami zachodzącymi w skali atomowej.
Wynik - jaki krakowscy fizycy osiągnęli we współpracy z warszawskimi Instytutami: Podstawowych Problemów Techniki PAN, a także Wysokich Ciśnień PAN - opublikowano w czasopiśmie „Journal of Materials Science”. O wynikach badań poinformowali przedstawiciele IFJ PAN w przesłanym PAP komunikacie.
"Staraliśmy się poznać na poziomie atomowym mechanizmy odpowiedzialne za obniżanie się prądu przebicia w kryształach węglika krzemu. Nasze obliczenia, wywodzące się z +pierwszych zasad+, prowadzą ku jakościowemu zrozumieniu problemu i przyczyniają się do wyjaśnienia szczegółów tego zjawiska” - mówi dr hab. Jan Łażewski, prof. IFJ PAN.
Pojęciem obliczeń „z pierwszych zasad” określa się obliczenia przeprowadzane z użyciem równań mechaniki kwantowej, wsparte jedynie wiedzą o budowie atomu i symetrii kryształów.
W podejściu tym nie ma żadnych bezpośrednich informacji z eksperymentów, co oznacza, że z jego pomocą można analizować również takie materiały, których jeszcze nikt nigdy nie badał, a nawet nie zsyntetyzował. Ze względu na dużą komplikację zagadnienia, do tej pory obliczenia z pierwszych zasad stosowano jedynie do zaburzeń punktowych, związanych z wakansami (brakami atomów, czyli dziurami w strukturze krystalicznej) lub domieszkami wprowadzanymi do kryształu.
Krakowscy badacze nie bez przyczyny zajęli się węglikiem krzemu. Właściwości tego półprzewodnika są tak interesujące, że kiedyś uważano go nawet za następcę krzemu. Kryształy węglika krzemu o niskiej jakości to popularny materiał ścierny. Materiał ten stosowany jest również w kamizelkach kuloodpornych i w tarczach hamulcowych najdroższych samochodów świata, takich jak Lamborghini czy Bugatti. Wysokiej jakości kryształy służą do wyrobu zwierciadeł teleskopów i elementów wysokonapięciowych urządzeń o dużej odporności na temperaturę.
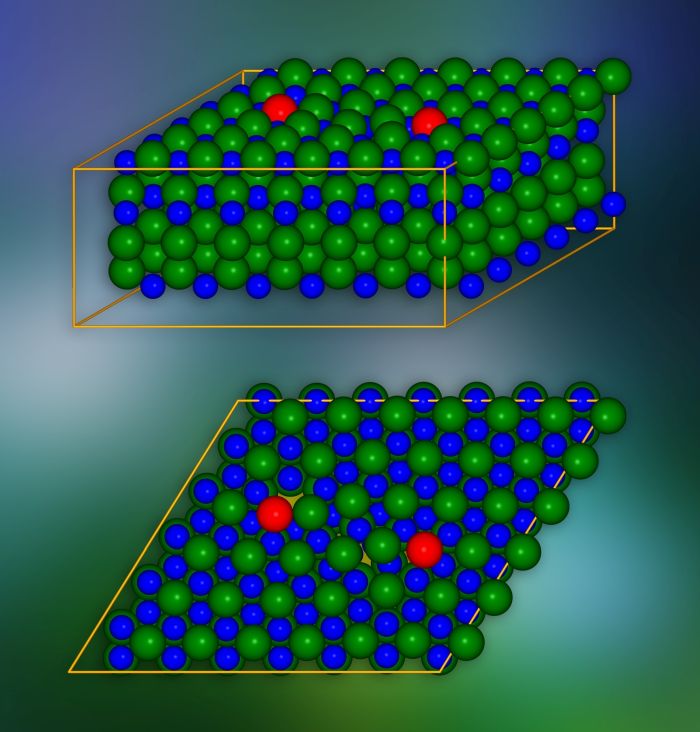
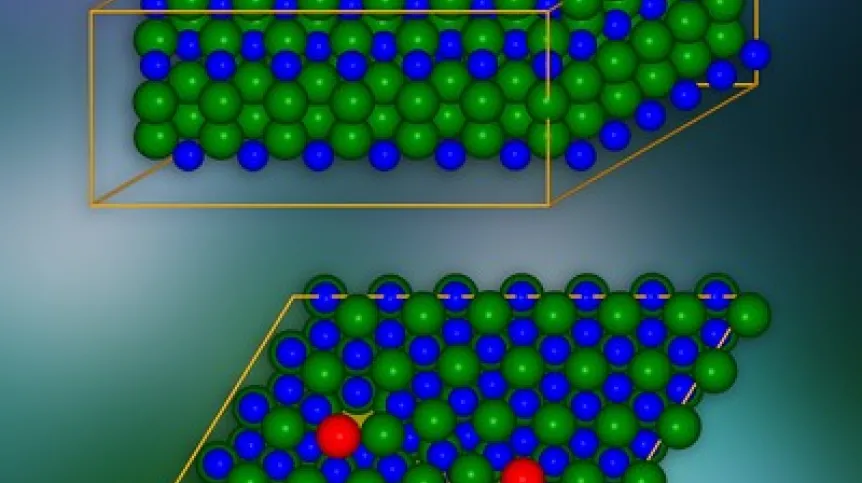
Model kryształu węglika krzemu z dyslokacjami krawędziowymi wprowadzonymi w miejscach zaznaczonych na czerwono. Na dole wygląd pojedynczej płaszczyzny krystalograficznej. Na żółto wyróżniono miejsca, przez które ładunki elektryczne mogą „wyciekać” do sąsiednich warstw. Źródło: IFJ PAN
Na poziomie atomowym kryształy węglika krzemu są zbudowane z wielu ułożonych jedna na drugiej płaskich warstw. Każda warstwa przypomina plaster miodu: składa się z sześciokątnych komórek, w których narożnikach są ulokowane pionowo cząsteczki węglika krzemu. Każde dwie sąsiednie warstwy można połączyć na trzy sposoby. Wielowarstwowe „kanapki” o różnych wzajemnych ułożeniach tworzą tzw. modyfikacje politypowe, których w przypadku węglika krzemu jest ponad 250. Grupa z IFJ PAN zajmowała się politypem oznaczonym jako 4H-SiC.
„Przy modelowaniu tego typu struktur jednym z podstawowych problemów jest złożoność obliczeniowa. Model kryształu czystego, pozbawionego domieszek czy dyslokacji, charakteryzuje się dużą symetrią i można go przeliczyć nawet w kilka minut. Żeby zrobić rachunek dla materiału z dyslokacją, potrzebujemy już całych miesięcy pracy komputera o dużej mocy obliczeniowej” - podkreśla dr hab. Paweł Jochym, prof. IFJ PAN.
Model kryształu SiC składał się z około 400 atomów. Przeprowadzone symulacje wykazały, że w warstwach kryształów, wzdłuż krawędzi rdzenia defektu, pojawiają się „tunele” w formie kanałów o zmniejszonej gęstości ładunku. Obniżają one lokalnie barierę potencjału i powodują, że ładunki elektryczne mogą „wyciekać” z pasma walencyjnego. Dodatkowo w przerwie wzbronionej, która w izolatorze gwarantuje brak przewodzenia prądu elektrycznego, pojawiają się stany redukujące jej szerokość i skuteczność w ograniczaniu przepływu ładunku. Wykazano, że stany te pochodzą od atomów ulokowanych w rdzeniu dyslokacji.
„Sytuację można porównać do głębokiego, stromego wąwozu, który próbuje pokonać wiewiórka. Jeśli dno wąwozu jest puste, wiewiórka nie przedostanie się na drugą stronę. Jeśli jednak na dnie rośnie pewna liczba odpowiednio wysokich drzew, wiewiórka może po ich wierzchołkach przeskoczyć na drugą stronę wąwozu. W modelowanym przez nas krysztale wiewiórką są ładunki elektryczne, pasmo walencyjne to jedna krawędź wąwozu, pasmo przewodnictwa – druga, a drzewami są wspomniane stany związane z atomami rdzenia dyslokacji” - mówi prof. Łażewski.
Teraz, gdy mechanizmy odpowiedzialne za obniżanie progu bariery energetycznej stały się znane na poziomie atomowym, pojawiło się ogromne pole do popisu dla eksperymentatorów. Zaproponowany mechanizm trzeba będzie zweryfikować, by później móc go użyć do ograniczenia negatywnego wpływu badanych defektów. Na szczęście istnieją już odpowiednie ku temu możliwości techniczne.
„Przyszłość zweryfikuje, czy nasze pomysły zostaną potwierdzone w całości. Jesteśmy jednak spokojni o losy naszego modelu i zaprezentowanego podejścia do symulowania dyslokacji krawędziowych. Już teraz wiemy, że model +z pierwszych zasad+ sprawdził się w konfrontacji z niektórymi danymi eksperymentalnymi” - podsumowuje prof. Jochym.
PAP - Nauka w Polsce
lt/ ekr/
Fundacja PAP zezwala na bezpłatny przedruk artykułów z Serwisu Nauka w Polsce pod warunkiem mailowego poinformowania nas raz w miesiącu o fakcie korzystania z serwisu oraz podania źródła artykułu. W portalach i serwisach internetowych prosimy o zamieszczenie podlinkowanego adresu: Źródło: naukawpolsce.pl, a w czasopismach adnotacji: Źródło: Serwis Nauka w Polsce - naukawpolsce.pl. Powyższe zezwolenie nie dotyczy: informacji z kategorii "Świat" oraz wszelkich fotografii i materiałów wideo.












